
An inflammasome represents a high molecular weight complex that activates inflammatory caspases and activates cytokines of the IL-1 family. Several inflammasomes have been described and were so far defined by the NLR protein that they contain – the NLRP1 (NALP1) inflammasome, the NLRP3 (NALP3) inflammasome and the IPAF (NLRC4) inflammasome. Interestingly, the AIM2 (absent in melanoma 2) inflammasome, which has been described recently, does not contain any member of the NLR family. Inflammasomes can be activated through multiple signals including live bacteria, microbial toxins, xeno-compounds, PAMPs and DAMPs. It is suggested that the LRR domains of NLRP3 mediate autorepression, probably by SGT1 and HSP90 chaperones that maintain NLRP3 in an inactive, but signal-competent state. Upon sensing of their respective ligands, NLRP1 or NLRP3 oligomerise via the NACHT domain, which leads to PYD clustering and to the recruitment of the adapter protein ASC (apoptosis-associated speck-like protein containing a CARD) due to homotypic PYD-PYD interactions. In the case of AIM2, oligomerisation is likely mediated by clustering upon multiple binding sites within dsDNA and not by a central oligomerisation domain like NACHT. Inflammasome ComplexHowever, AIM2 oligomerisation also leads to the recruitment of ASC via homotypic PYD-PYD interactions. ASC contains an N-terminal PYD domain and a C-terminal CARD domain that allows the 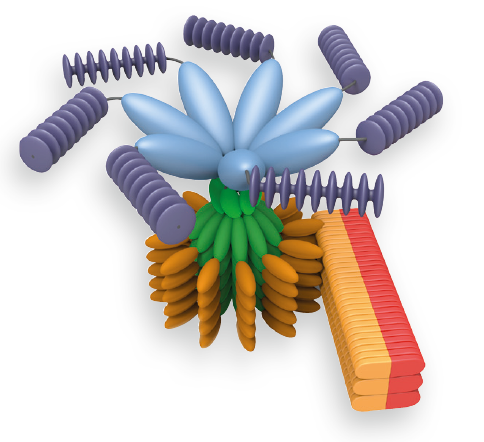 recruitment of inflammatory caspases through homotypic CARD-CARD interactions. Thus inflammatory caspases are brought into close proximity which permits autoactivation and formation of the active caspase. In the case of procaspase-1, a p10/p20 tetramer is formed after autocleavage. In addition to caspase-1, NLRP1 can also recruit caspase-5 to the complex but the role of caspase-5 is still under debate. Contrary to NLRP1, NLRP3 and AIM2, IPAF does not recruit an adapter molecule but directly interacts with procaspase-1 via its CARD domain (see Figure below). However, depending on the IPAF stimulus, maximal caspase-1 activation downstream of IPAF can require ASC or NAIP. The assembly of the different inflammasomes elicits a common downstream cascade, namely the activation of inflammatory caspases. These include caspase-1, -4 and -5 in humans and caspase-1, -11 and -12 in mice. However, caspase-1 appears to be the most dominant inflammatory caspase associated with inflammasomes. The inflammatory caspases all have a CARD domain followed by a domain containing the catalytic residue cysteine and are called inflammatory caspases because their main substrates are cytokines (such as pro-IL-1β, pro-IL-18 and eventually pro-IL-33) which are cleaved to their active and secreted form. In addition, inflammasome activation can lead to host cell death in certain cell types, known as pyroptosis. Pyroptosis is thought to be important in restricting the intracellular replication of invasive pathogens.
recruitment of inflammatory caspases through homotypic CARD-CARD interactions. Thus inflammatory caspases are brought into close proximity which permits autoactivation and formation of the active caspase. In the case of procaspase-1, a p10/p20 tetramer is formed after autocleavage. In addition to caspase-1, NLRP1 can also recruit caspase-5 to the complex but the role of caspase-5 is still under debate. Contrary to NLRP1, NLRP3 and AIM2, IPAF does not recruit an adapter molecule but directly interacts with procaspase-1 via its CARD domain (see Figure below). However, depending on the IPAF stimulus, maximal caspase-1 activation downstream of IPAF can require ASC or NAIP. The assembly of the different inflammasomes elicits a common downstream cascade, namely the activation of inflammatory caspases. These include caspase-1, -4 and -5 in humans and caspase-1, -11 and -12 in mice. However, caspase-1 appears to be the most dominant inflammatory caspase associated with inflammasomes. The inflammatory caspases all have a CARD domain followed by a domain containing the catalytic residue cysteine and are called inflammatory caspases because their main substrates are cytokines (such as pro-IL-1β, pro-IL-18 and eventually pro-IL-33) which are cleaved to their active and secreted form. In addition, inflammasome activation can lead to host cell death in certain cell types, known as pyroptosis. Pyroptosis is thought to be important in restricting the intracellular replication of invasive pathogens.
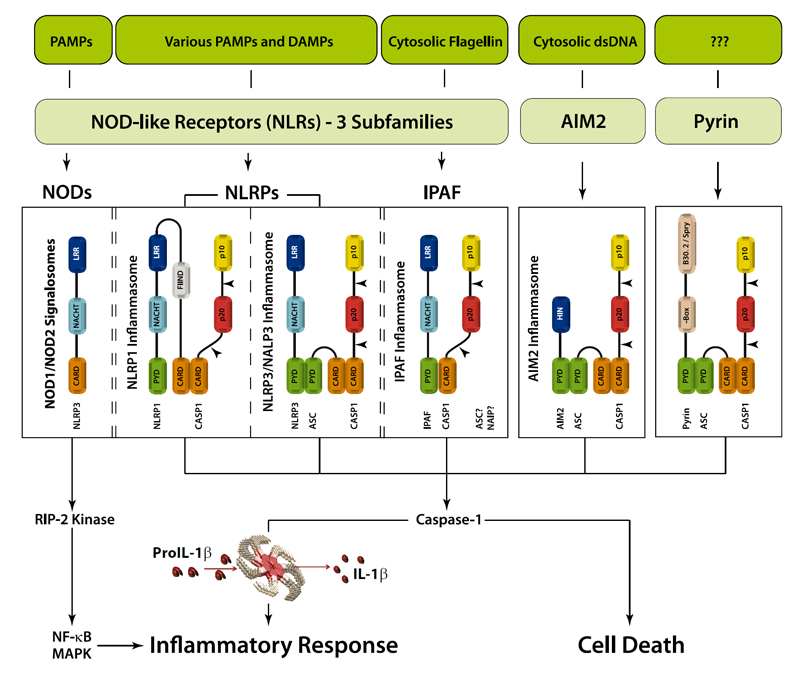
Figure: Danger signals or bacterial compounds as activators of inflammasomes and NODs.
Inflammasomes − Activity Regulation
Even though mechanisms that regulate inflammasome activity remain elusive, various proteins were identified that may interfere with inflammasome activation and inflammasome-dependent inflammatory caspase processing. In general, two subtypes of inflammasome regulators can be distinguished – those containing a CARD domain and those with a PYD domain. Such proteins encompass not only host-derived inflammasome regulators but also various bacterial virulence factors inhibiting caspase-1 activation and viral PYD proteins.
Inflammasomes − Therapeutic Implications
As IL-1β and other cytokines are key players in the inflammatory response, it is tempting to speculate that IL-1β, inflammatory caspases and inflammasomes play an important role in several diseases (see Figure below). Indeed, some human hereditary or acquired diseases have been linked to elevated IL-1β, some of which can be treated by antagonists against IL-1β or its receptor. A number of diseases, known as cryopyrin-associated periodic syndromes (CAPS), have been directly linked to NLRP3 mutations. Furthermore, gout, an autoinflammatory disease characterised by severe joint inflammation, is associated with the deposition of MSU crystals in joints, among other characteristics. As MSU is a potent NLRP3 inflammasome agonist, it is believed that inflammasome-regulated IL-1β exerts a pathogenic role in gout. Furthermore, IL-1β secretion by the NLRP3 inflammasome is triggered by high extracellular glucose in b-cells. Elevated IL-1β is a risk factor for the development of Type 2 Diabetes Mellitus (T2DM) and contributes to insulin resistance. Thus by functioning as a sensor for metabolic stress, like in the form of monosodium urate (MSU) or hyperglycemia, the NLRP3 inflammasome likely contributes to the pathogenesis of gout or T2DM, respectively. In addition, several inflammasome regulators have significant relevance in diseases. In patients with FMF (familial Mediterranean fever), pyrin was shown to be mutated. Elevated IL-1β levels in the autoinflammatory disease PAPA (pyogenic arthritis, pyoderma gangrenosum, and acne) are associated with mutations in PSTPIP1, a pyrin-interacting protein. Together, this indicates the importance of pyrin and inflammasome regulators in autoinflammatory diseases and may allow novel entry points for disease treatment.
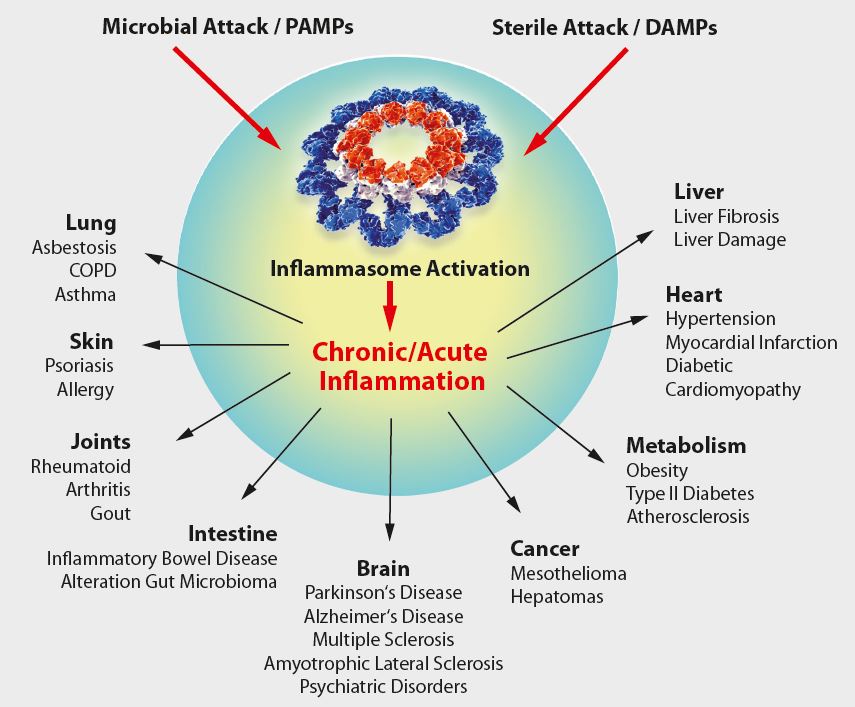
Figure: Overview of inflammasome-associated diseases.
Detailed View on the Inflammasomes & Their Signaling
NLRP1 Inflammasome
The anti-apoptotic proteins Bcl-2 and Bcl-XL were recently shown to bind and inhibit NLRP1. Bcl-2 and Bcl-XL inhibit ATP binding to NLRP1, which is required for oligomerisation and furthermore, Bcl-XL was shown to suppress NLRP1 oligomerisation. Thus inflammatory caspases are not brought into close proximity which negatively interferes with caspase-1 autoactivation and subsequent processing of pro-inflammatory cytokines. In addition to those potential NLRP1 activity regulators, K+ efflux appears to be essential for NLRP1 activation.
NLRP3/NALP3 Inflammasome
Even though the exact mechanism of NLRP3/NALP3 inflammasome activation is still under debate, three models are favoured among researchers:
1) The NLRP3 inflammasome agonist, extracellular ATP, binds to its receptor P2X7 what triggers K+ efflux and pannexin-1 membrane pore formation. The latter may enable the entry of extracellular factors, which can be direct NLRP3 activators. It is also suggested that NLRP3 may sense the K+ efflux or membrane integrity.
2) Phagocytosed crystalline or particulate NLRP3 ligands, such as MSU, alum, silica and asbestos, can cause lysosomal destabilisation and rupture due to mechanical insult which leads to the release of lysosomal content into the cytosol. As this pathway is sensitive to the cathepsin B inhibitor CA-074-Me, it was suggested that cathepsin B, a lysosomal protease, is involved in the activation of a direct NLRP3 ligand. However, no altered IL-1β secretion and caspase-1 cleavage in response to NLRP3 ligands, like MSU or alum, was observed in cathepsin B deficient macrophages. It is yet not known how NLRP3 senses cytoplasmic lysosomal content.
3) The third model involves the production of reactive oxygen species (ROS). All NLRP3 agonists tested lead to the production of ROS and furthermore, ROS scavengers suppress NLRP3 activation. The cellular source of ROS is currently unknown and although ROS seems to be necessary for NLRP3 inflammasome activation, it is not sufficient. In B cells, TXNIP (thioredoxin-interacting protein; VDUP1), which is a ROS-sensitive NLRP3 ligand, was shown to be involved in triggering NLRP3 activation.
Another important parameter in NLRP3 activation, as for NLRP1, seems to be the cytoplasmic K+ concentration. The fact that macrophages cultured in high K+ concentration show a decreased NLRP3-dependent caspase-1 activation likely implies that K+ efflux is necessary upstream of NLRP3 activation.
IPAF Inflammasome
The IPAF inflammasome becomes activated by gram-negative bacteria possessing a functional T3SS or T4SS, like S. typhimurium, S. flexneri, L. pneumophila, and P. aeruginosa. Initially, cytosolic flagellin was shown to trigger the IPAF inflammasome activity. However, as non-flagellated bacteria like S. flexneri also induce IPAF inflammasome activation, it is very likely that there exist other IPAF agonists. As NLRP3, IPAF was shown to bind SGT1 and HSP90. The inhibition of HSP90 by geldanamycin blocks the IPAF activity, indicating that HSP90 is somehow crucial for IPAF signalling. Unlike NLRP1 and NLRP3, IPAF activation is not inhibited by high extracellular K+ concentration, indicating that IPAF is not a sensor for ionic fluxes. However, no direct ligand-receptor interaction was observed to date.
AIM2 Inflammasome
The HIN-200 family member, AIM2, acts as a sensor for cytosolic bacterial, viral and host dsDNA and triggers an inflammatory response through the formation of the AIM2 inflammasome. It is suggested that AIM2 binds directly to dsDNA via its C-terminal HIN-200 domain. As AIM2 recognises host dsDNA it might be involved in autoimmune diseases. The discovery of the AIM2 inflammasome is not only remarkable because AIM2 is the first non-NLR family member forming an inflammasome, but also because AIM2 is the first inflammasome receptor shown to directly interact with its ligand.
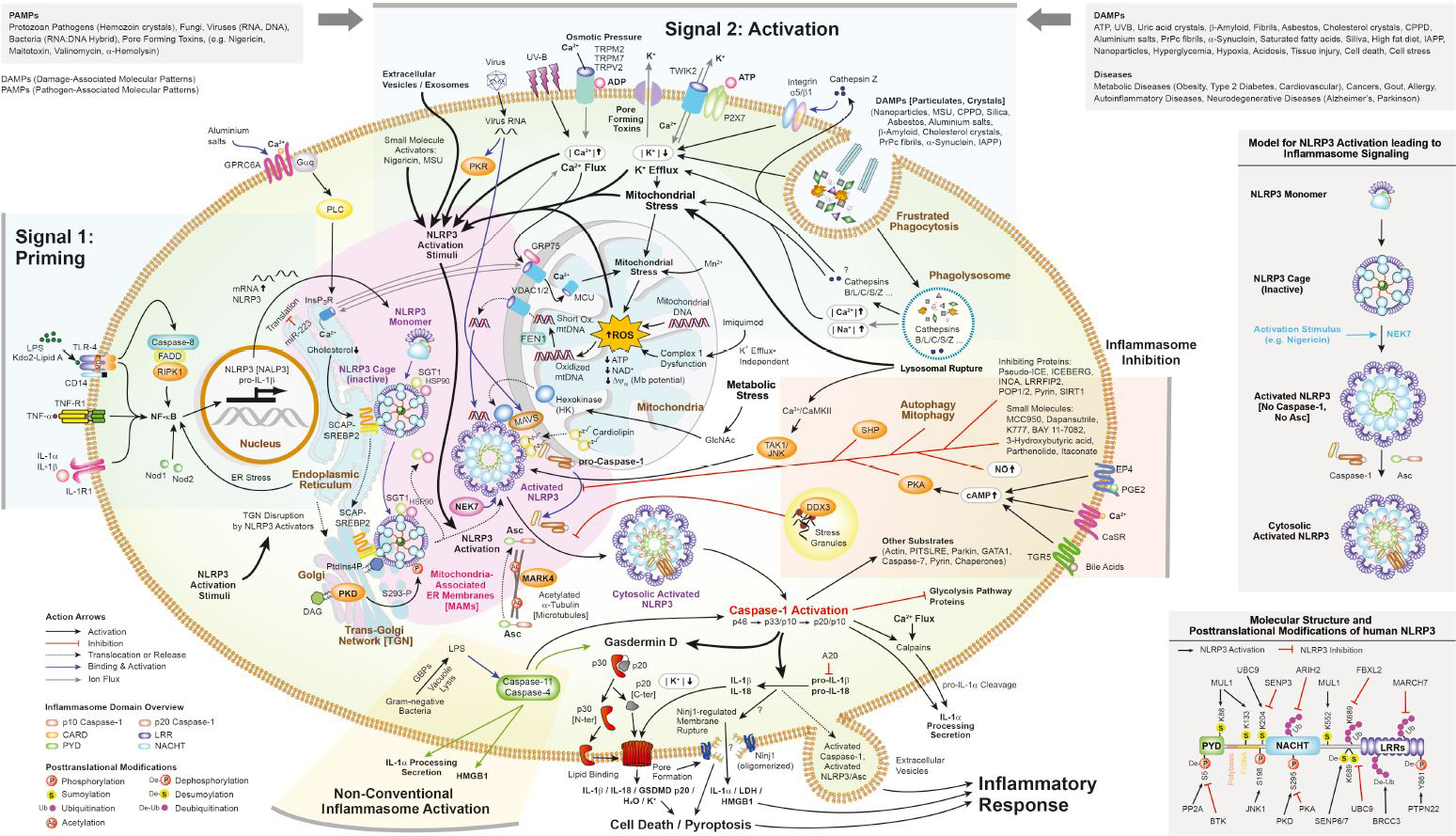
Figure: Inflammasome Signaling
Measuring the Inflammasomes
Inflammasomes are multi-protein complexes implicated in physiological and pathological inflammation. Immunoblotting for caspase-1 is the gold-standard method of detecting inflammasome activation, but also presents certain technical difficulties, particularly with the choice of protein precipitation, protein separation and transfer protocols. Priming and Activation steps are crucial for successful immunoblotting as well. Listed below are Spinger Protocols from the Series of Methods and Protocols using Adipogen’s Standard Antibodies for Inflammasomes detection and Tipps for NLRP3 Western Blotting.
NLRP3 Inflammasome Starter Sets
|
PID |
PRODUCT NAME |
PROTOCOL |
| NLRP3 Inflammasome Human Antibodies Starter Set Manual |
The NLRP3 Inflammasome Human Antibodies Starter Set is an all-in-one solution to study the NLRP3 inflammasome using Western blotting application. This antibody starter set comprises our KO extract validated STANDARD antibodies against the key components of the NLRP3 inflammasome including NLRP3, Asc and Caspase-1, used and published by the experts in inflammasome research. This economic starter set contains enough primary antibodies to perform at least 3 western blot experiments. |
|
| NLRP3 Inflammasome Mouse Antibodies Starter Set Manual |
The NLRP3 Inflammasome Mouse Antibodies Starter Set is an all-in-one solution to study the NLRP3 inflammasome using Western blotting application. This antibody starter set comprises our KO extract validated STANDARD antibodies against the key components of the NLRP3 inflammasome including NLRP3, Asc and Caspase-1, used and published by the experts in inflammasome research. This economic starter set contains enough primary antibodies to perform at least 3 western blot experiments. |
|
| NLRP3 Inflammasome Human Reagents Starter Set Manual |
The NLRP3 Inflammasome Human Reagents Starter Set is an all-in-one solution to study the NLRP3 inflammasome using Western blotting application. This reagents starter set comprises our KO extract validated STANDARD antibodies against the key components of the NLRP3 inflammasome including NLRP3, Asc and Caspase-1, used and published by the experts in inflammasome research. It also contains two chemical compounds for priming and activation of the NLRP3 inflammasome. This economic starter set contains enough primary antibodies to perform at least 3 western blot experiments. |
|
| NLRP3 Inflammasome Mouse Reagents Starter Set Manual |
The NLRP3 Inflammasome Mouse Reagents Starter Set is an all-in-one solution to study the NLRP3 inflammasome using Western blotting application. This reagents starter set comprises our KO extract validated STANDARD antibodies against the key components of the NLRP3 inflammasome including NLRP3, Asc and Caspase-1, used and published by the experts in inflammasome research. It also contains two chemical compounds for priming and activation of the NLRP3 inflammasome. This economic starter set contains enough primary antibodies to perform at least 3 western blot experiments. |
THE STANDARD KO-extract validated Antibodies – Used and published by the Experts!
| PID | PRODUCT NAME | PROTOCOL |
|
anti-Caspase-1 (p20) (mouse), mAb (Casper-1) |
Measuring the Inflammasome (Download Protocol) Immunoblotting for Active Caspase-1 (Download Protocol) Inflammasome Activation and Inhibition in Primary Murine Bone Marrow-Derived Cells, and Assays for IL-1α, IL-1β, and Caspase-1 (Download Protocol) Assessing Caspase-1 Activation (Download Protocol) |
|
|
anti-Caspase-1 (p10) (mouse), mAb (Casper-2) |
Measuring the Inflammasome (Download Protocol) |
|
|
anti-Caspase-1 (p20) (human), mAb (Bally-1) |
Measuring the Inflammasome (Download Protocol) Cell-Free Assay for Inflammasome Activation (Download Protocol) |
|
|
anti-NLRP3/NALP3, mAb (Cryo-2) |
Measuring NLR Oligomerization I: Size Exclusion Chromatography, Co-immunoprecipitation, and Cross-Linking (Download Protocol) Functional Reconstruction of NLRs in HEK293 Cells (Download Protocol) |
|
|
anti-NLRP1b (mouse), mAb (2A12) |
Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes: A. Sandstrom, et al.; Science 364, 82 (2019) (Publication) |
|
|
anti-Asc, pAb (AL177) |
Measuring Inflammasome Activation in Response to Bacterial Infection (Download Protocol) Measuring NLR Oligomerization II: Detection of ASC Speck Formation by Confocal Microscopy and Immunofluorescence (Download Protocol) Cell-Free Assay for Inflammasome Activation (Download Protocol) Flow Cytometric Detection of AC Specks (Download Protocol) |
|
|
anti-RIG-I, mAb (Alme-1) |
The Human Papillomavirus E6 Oncoprotein Targets USP15 and TRIM25 To Suppress RIG-I-Mediated Innate Immune Signaling: C. Chiang, et al.; J. Virol. 92, e01737-17 (2018) [KO Validation] (Publication) |
|
|
anti-ZBP1, mAb (Zippy-1) |
Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1: H. Guo, et al.; Cell Death Dis. 9, 816 (2018) [KO Validation] (Publication) |
Tips for NLRP3 Western Blotting!
i) Cells or Tissues:
– Verify in the literature that primary cells, tissues or cell lines used express NLRP3 at detectable levels. Priming with LPS, PMA or other priming reagents (that activate NF-κB signaling) can be performed to increase NLRP3 levels.
– NLRP3 is expressed in many different cells or tissues, but in some cases (unexpected mutations in some cell subclones, others), levels of NLRP3 can be regulated leading to no or weaker signal in WB.
ii) SDS-PAGE gel:
– Percentage SDS-PAGE should be optimally <8%.
– All buffers (running, stacking, transfer) should be made fresh.
– Load at least 20-30µg per lane.
– Transfer is important since NLRP3 is a tricky protein not so easy to transfer.
– Transfer should be performed in a wet tank device overnight at 20V at 4°C .
– No transfer for 1h and also dry or semi-dry devices are NOT recommended.
iii) Primary antibody:
– Use freshly prepared antibody anti-NLRP3 (Cryo-2) at a dilution of 1/1’000. If protein is weakly expressed, a lower dilution, such as 1/500 can be used.
iv) Secondary antibody:
– Use a freshly prepared anti-mouse IgG (AG-29B-0004E) antibody coupled to HRP (1/5’000) (or recommended dilution mentioned by the manufacturer if a different secondary anti-mouse IgG-HRP is used).
v) Positive controls:
– As positive control cell lines (that can be primed with ATP, PMA or LPS to increase NLRP3 levels), we recommend human monocyte-like cells, such as THP1 or U937 and mouse macrophage-like cells Raw264.7 or mouse monocyte /macrophage cell J774A.1.
Notes:
- Adipogen anti-NLRP3 antibody (Cryo-2) is one of the very few NLRP3 antibody validated using KO extracts. Many antibodies on the market are not validated and have been shown to be not specific.
- Adipogen anti-NLRP3 antibody (Cryo-2) detects the N-terminus Pyrin domain.
Co-culture protocol to study in vitro the role of inflammasomes in myeloid cells (macrophages) on T cells down-regulation
A key observation of tissue injury, such as stroke and burn, is a state of systemic immunosuppression characterised by loss of T cells and the rise of infections. J. Zhu, et al. (2021) present an in vitro model for cell-cell interactions between innate (macrophages) and adaptive (T cells) immune cells. This protocol facilitates bone marrow-derived macrophages (BMDMs) and splenic T cells in a co-culture model. The procedure mimics injury-induced T cell death, which is driven by inflammasome activation in macrophages.
LIT: A macrophage-T cell coculture model for severe tissue injury-induced T cell death: J. Zhu, et al.; STAR Protocols 2, 100983 (2021) | Post-injury immunosuppression and secondary infections are caused by an AIM2 inflammasome-driven signalling cascade: S. Roth, et al.; Immunity 54, 648 (2021)
The Key Inflammasome Activators and Inhibitors
| Product Name | PID | Product Description |
| Monosodium urate (crystals) Monosodium urate (ready-to-use) | AG-CR1-3950 AG-CR1-3951 | Potent NLRP3 inflammasome activator. |
| Nigericin . Na | AG-CN2-0020 | Potent NLRP3 inflammasome activator. |
| N-Acetyl-D-glucosamine | AG-CN2-0489 | Acts as activator of NLRP3 inflammasome by dissociating the enzyme hexokinase from the mitochondria. |
| Necrosulfonamide | AG-CR1-3705 | Binds directly to Gasdermin D and inhibits the oligomerization of the N-terminus and therefore the pore formation and pyroptosis. |
| U-73122 | AG-CR1-3698 | Gasdermin D N-terminal fragment (GSDMD-N)-induced pyroptosis inhibitor. Protects against GSDMD-N cytotoxicity in macrophages or against lethal infection in mice. |
| Dapansutrile | AG-CR1-3535 | Potent, selective and orally active NLRP3 inflammasome inhibitor. Directly binds to the ATP-binding motif of NLRP3 NACHT domain and inhibits NLRP3 inflammasome assembly and activation. |
| MCC950 . Na (water soluble) | AG-CR1-3615 | Potent and selective NLRP3 inflammasome inhibitor. |
| Isoliquiritigenin | AG-CN2-0459 | Inhibits NLRP3-activated Asc oligomerization. Blocks priming and activation steps. |
| BAY 11-7082 | AG-CR1-0013 | Reduces ATPase activity of the NLRP3 inflammasome. |
| (R)-3-Hydroxybutyric acid (S)-3-Hydroxybutyric acid DL-3-Hydroxybutyric acid sodium salt | AG-CR1-3616 AG-CR1-3617 CDX-H0080 | Prevent K+-efflux and consequently reduce Asc oligomerization and speck formation. |
| K777 [K11777] | AG-CR1-0158 | Broad-range cathepsin inhibitor useful for inflammasome inhibition. |
| m-3M3FBS | AG-CR1-3548 | Triggers mitochondrial damage and NLRP10 inflammasome activation. |
Priming Step of Inflammasome Activation
The most prominent function of the NLRP3 inflammasome is the processing and activation of pro-interleukin-1β (pro-IL-1β). Yet most cells do not express pro-IL-1β and thus prior expression of pro-IL-1β is required. This can be achieved by stimulating receptors such as TLRs (e.g. through LPS or PMA), Nods, TNF-Rs (e.g. through TNF-α) or IL-1R1 (through IL-1α and IL-1β) that activate NF-κB and initiate pro-IL-1β transcription. This process is called priming. Priming also induces NF-κB-dependent transcription of NLRP3.
Originally posted on adipogen.com/inflammasomes
Caltag Medsystems is the distributor of Adipogen products in the UK and Ireland. If you have any questions about these products, please contact us.